

18 min read
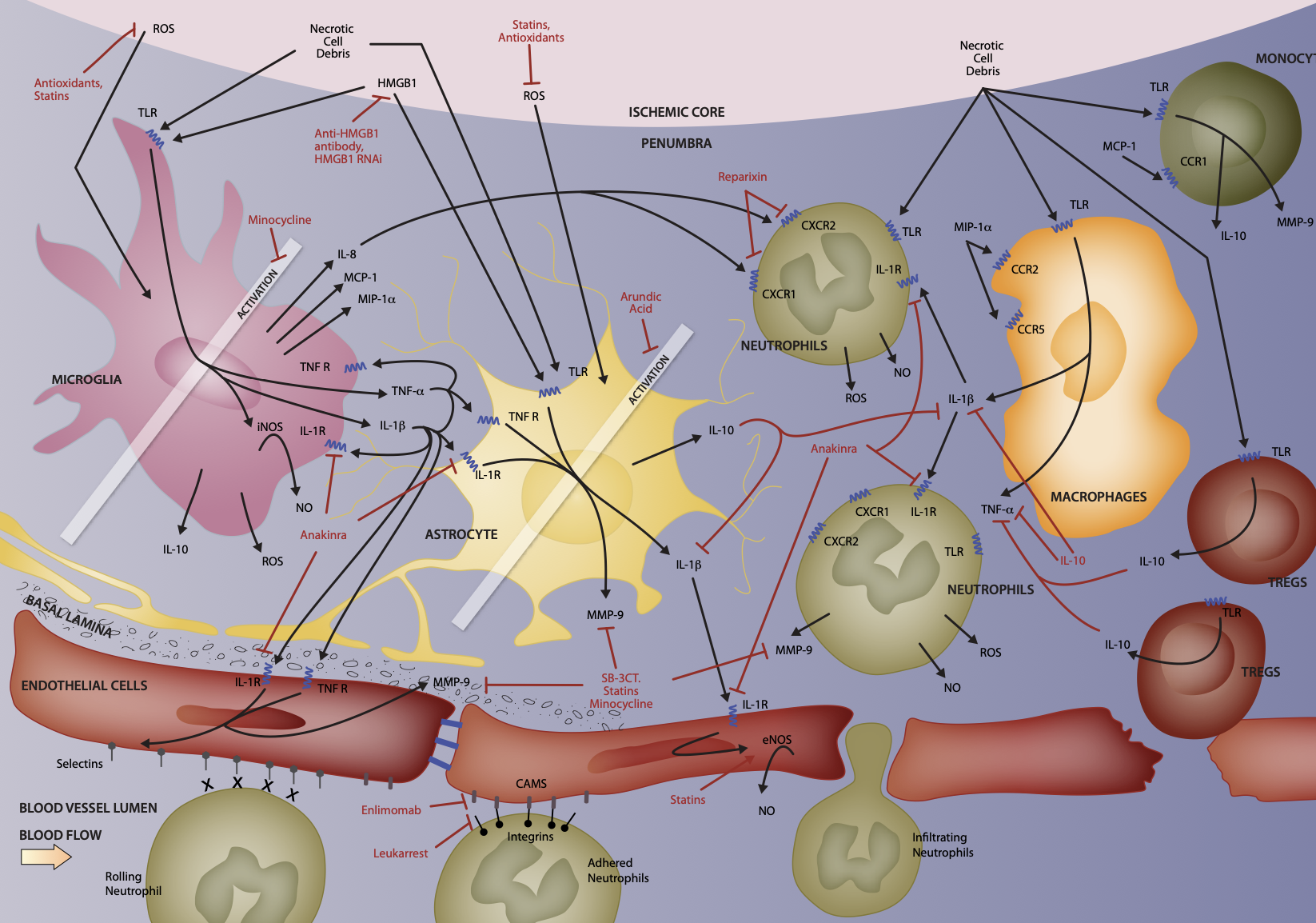
Cellular and molecular interactions involved in neuroinflammation. Potential targets for improved patient outcome.
The most common form of stroke is acute isch- emic stroke (approximately 85% of cases), which is caused by either an atherothrombosis in a major cervical or intracranial artery or an embolism that travels from the heart. The resulting occlusion causes a sudden deficiency of oxygen and glucose in the brain region nor- mally serviced by the blocked artery. Victims of large-vessel ischemic strokes lose on the order of 100 million neurons per hour prior to treatment, causing immediate, permanent neural damage in the infarct area, termed the ischemic core. Further neural damage occurs in the areas surrounding the core, called the penumbra, where the tissue becomes highly inflamed and slowly dies. Stroke sufferers experience a range of neurological deficits including partial paralysis, impaired memory, loss of speech, and/or decreased cognition and many become permanently disabled, requiring institutional care. [1-4]
Unfortunately, current stroke therapies approved for human use are very limited. The only drug clinically available in the US is intrave- nous recombinant tissue plasminogen activator (rt-PA),athrombolyticagentthathasbeenshown to improve stroke patient functional outcomes. However, rt-PA is only effective if administered within the first 3 hours after symptom onset and carries with it a significant risk of intracranial hemorrhage. Consequently, only about 5 to 10% of patients can receive this therapy. Over 1,000 other potential stroke therapeutics have been tested in animals and approximately one tenth of these have made it to clinical trials. However, the majority of these efforts have already failed. It is likely that the most effective way to improve outcomes is rapid reperfusion of the ischemic area using thrombolytic means in combination with neuroprotective strategies to salvage cells within the penumbra and prevent them from becoming part of the ischemic core. To this end, researchers have been increasingly focused on post-stroke neuroinflammation and the role it plays in neurotoxicity and neuroprotection. [1-4]
Post-stroke neuroinflammation is a very complex phenomenon involving multiple resident and invading cell types at varying degrees of differentiation or activation each expressing specific subsets of diffusible factors, receptors, cellular adhesion molecules, and other markers, all of which is chang- ing as time passes to create an initially neurotoxic and then finally neuroprotective environment. This inflammatory process in the penumbra offers a broad array of potential cellular and molecular targets with much wider therapeutic windows. At the cellular level, neurons, microglia, astrocytes, and cere- brovascular endothelial cells are the first affected by the ischemic conditions and their responses to massive cell death in neighboring tissue initiates the precisely timed arrival of successive subsets of leukocytes – first neutrophils, followed by monocytes and macrophages, and finally T cells. Targeting these cells via manipulation of their phenotypes or activation states or their movements into lesion sites or their release of harmful mediators represents a major investigative pathway toward potential therapeutics for ischemic stroke sufferers. [1-4]
Microglia:
Microglia are the first to detect and respond to ischemic conditions, showing signs of activation as early as a few minutes after infarct. Microglia are the resident brain immunocompetent cells and constitutively express a wide variety of pattern recognition receptors. After stroke, resting microglia detect the spilled cytoplasmic contents of necrotic cells as well as high levels of ROS and consequently transform to an active, phagocytic phenotype, functioning to clear dead cells and cellular debris and release pro-inflammatory and cytotoxic diffusible factors. Inhibitors of microglial activation, such as the multi-function- al drug Minocycline, have been shown to reduce infarct size in animal models when administered intraveneously in combination with rt-PA within 6 hours of infarct. At later timepoints, it appears microglia have neuroprotective effects, such as producing neurotrophic and other growth factors and supporting neurogenesis and neural plasticity. [1-4]
Astrocytes:
Astrocytes normally provide energy, neu- rotrophic and neurogenic factors, neurotrans- mitter precursors, and anti-oxidant defense to neurons. Like microglia, astrocytes also proliferate and differentiate in response to stroke and begin to produce pro-inflammatory and cytotoxic mediators and contribute to the loss of blood-brain barrier (BBB) integrity. Astroglial activity begins at the core approximately 4 hours after stroke and may be observed for up to 28 days. Modulators of astrocyte function such as Arundic Acid appear to improve outcomes in experimental animal models. Like microglia, astrocyte activity appears to be initially damag- ing and later protective. [2,4]
Endothelial Cells:
The BBB is composed of endothelial cells joined by tight junctions, a basal lamina of extracellular matrix and the endfeet of astrocytes and is meant to protect and maintain the brain’s delicate extracellular microenvironment. Breakdown of the BBB is a pathological hallmark of ischemic stroke and significantly contributes to resulting brain damage and poor outcome. Upon ischemic insult, the BBB becomes disrupted and hyperpermeable in a biphasic pattern with a first opening at 2 to 3 hours and a second opening at 24 to 48 hours post-stroke. Depending on the severity of the BBB dysfunction, endothelial cells may suffer edema, astrocytes may detach, and whole vessels may rupture. Prevention of the opening of the BBB after stroke is a major therapeutic goal. [1-3]
Neutrophils:
Neutrophils are the first of the leukocyte subtypes to arrive on the scene, where they are detectable within a few minutes of trauma. Neutrophils adhere to and then migrate through the endothelial vessel walls and subsequently follow chemical gradients toward damaged brain tissue. Neutrophils remain within the brain for up to 15 days where they contribute to neu- roinflammation by releasing pro-inflammatory molecules and cytotoxic factors and promote the disruption of the BBB by producing proteas- es. While neutrophils do assist by phagocytos- ing damaged cells and debris, they are generally thought to have overall deleterious effects on penumbral tissue. Strategies for pharmacological inhibition of neutrophil chemotaxis, infiltration, and degranulation are being investigated. However, therapeutic attempts to in some way prevent neutrophil activity after stroke have met with mixed results so far. [2-4]
Monocytes and Macrophages
Peripheral blood monocytes arrive at the injury site approximately 4 to 6 hours after neutrophils. They leave circulation and enter the brain by crossing the now-damaged BBB. Once infiltrated into brain tissue, these cells phagocytose compromised neurons and glia, extracellular matrix debris, and spent neutrophils. Macrophages, like neutrophils, also act to amplify the inflammatory cascade in the core and the penumbra by similarly releasing pro-inflammatory and cytotoxic mediators. [2,3]
T Cells
T lympohocytes are attracted by the inflammatory environment in the core and penumbra and generally act as would be expected based on their phenotype. While cytotoxic T cells attack and kill both damaged and healthy cells and Th1 cells and γδT cells promote inflammation by further release of pro-inflammatory cytokines, Th2 cells and regulatory T (Treg) cells release anti-inflammatory mediators. Attempts to artificially influence specific T cell subtypes have also had mixed results. Perhaps the most promising cellular target is Treg cells. Animals lacking functional, circulating Treg cells or depleted of Treg cells suffer more significant brain damage and disabilities after stroke. Treg cells are thought to exert immunosuppressive effects after stroke, with some evidence suggesting that Treg cells may play an important part in preventing secondary development of autoimmunity. For example, brain-specific antibodies are detectable in humans after stroke, astrocytes are known to drive T cells to a Treg phenotype to limit autoimmunity in experimental animal models of multiple sclerosis, and concurrent or subsequent infection may potentiate the development of autoimmunity after stroke. [3,4]
At the molecular level, the complex web of diffusible bioactive compounds and their receptors and signaling cascades, precisely regulated in space (i.e., different cell types) and time (i.e., after onset of ischemic stroke) is on the one hand a curse as there will likely be no universal “cure” for ischemic stroke, but on the other hand a blessing as it provides innumerable opportunities for targeted therapeutic intervention. [1-4] A selection of these potential therapeutic targets is described here.
Reactive Oxygen Species (ROS)
The most immediate initiators of neuroinflammation are ROS. As mentioned earlier, ischemic conditions in the brain generate a variety of free
radical species, which have many serious, directly deleterious effects on exposed cells. In addition, ROS contribute to secondary injury by initi- ating inflammation. ROS induce the synthesis of several transcription factors including NF-κB and STAT3, which then result in the up-regulation of pro-inflammatory genes in neurons and glia as well as adhesion molecule genes in endothelial cells. ROS represent a major potential therapeutic target for improving stroke outcome and compounds possessing antioxidant properties such as Ebselen, Resveratol, and natural Phy- toalexin (found in grapes and red wine) appear to decrease stroke-induced brain damage in animals. Further, elevating endogenous antioxidants via activating genes that contain antioxidant response elements within their promoters results in improved free radical scavenging and maintenance of redox potential. In in vitro mod- el systems as well as in vivo animal models of stroke, up-regulation of these genes appears to be highly neuroprotective. Finally, manipulation of the three enzymes involved in the production of nitric oxide (NO), namely inducible NO synthase (iNOS), neuronal NO synthase (nNOS), and endothelial NO synthase (eNOS), has been investigated. So far, results show that inhibition of either iNOS or nNOS is neuroprotective and that eNOS activity after stroke is actually beneficial as it promotes the maintenance of the cerebral vasculature. Statin drugs, perhaps best known for their effects on cholesterol syn- thesis and circulating LDL clearance, have also been demonstrated improve stroke outcomes in animal models. Statins have a broad range of actions including functioning as antioxidants, up-regulating eNOS, and promoting angiogenesis. [1,2,4]
High-Mobility Group Box Protein Family Member 1 (HMGB1)
The other primary initiator of post-stroke neuroinflammation is necrotic cell debris. Large quantities of a broad variety of molecules normally confined to the intracellular space are released when rapid and wide-spread necrotic cell death occurs in the ischemic core. Neighboring cells in the penumbral region become bathed in this soup of debris, which contains several specific molecules that, when detected by intact cells serve as danger signals – for example, hyaluronic acid, mRNA, heat shock proteins, and others. [3] One such molecule, high-mobility group box protein family member 1 (HMGB1), has received particular attention in the context of stroke-related neuroinflammation. HMGB proteins are constitutively expressed by many cell types and function to directly bind DNA or histones and transcrip- tion factors thereby modifying macromolecu- lar structures and influencing DNA repair and transcription. HMGB1 is released passively as a result of necrosis and actively in response to apoptotic cell phagocytosis. HMGB1 itself acts as a pro-inflammatory cytokine in the context of stroke, inducing microglial activation and iNOS, IL-1β, and TNF-α expression in vitro and elevating iNOS and IL-1β expression in vivo. Targeting HMGB1 in animals models of stroke by RNA interference or neutralizing antibodies results in inhibition of microglial activation, suppression of neuroinflammation, and reduction of infarct volume. [2]
Toll-Like Receptors (TLRs)
Signals of cellular stress, such as HMGB1, are detected via cell surface pattern recognition receptors of various kinds including the toll-like receptor (TLR) family members. Although TLR expression patterns vary by species and cell type, generally TLR2 and TLR4 appear to be the most highly expressed family members in brain tissues and likely mediate immediate detection of necrotic cell debris, microglial activation, and initiation of the inflammatory cascade. TLR2 has been directly implicated in this process as pre- conditioning with a TLR2 agonist to create tolerance results in reduced ischemic brain damage. All TLR family members are expressed on the surface of leukocytes, with varying subsets on different cell types. TLRs are known to function as monomers or homo- or heterodimers, changing their association with different adaptor proteins and MyD88-dependent and/or -independent signal transduction cascades, which ultimately influence transcription factors including NF-κB, AP1, c-JUN, and others that control transcrip- tion of inflammatory mediators. TLRs represent a tremendous opportunity for pharmacological manipulation of the ensuing neuroinflammatory cascade. However, it is only relatively recently that TLRs have been discovered and even more recently that they have been implicated in stroke, so these potential targets are still early in their development. [3]
IL-1β
IL-1β is released primarily by activated microglia, but also by astrocytes, neurons, and endothelial cells in the early stages following stroke and then by infiltrating leukocytes. Elevated IL-1β protein levels in the brain are detectable within a few hours after stroke and remain high for up to 4 days. IL-1β is an endogenous pyrogen and a strong pro-inflammatory cytokine that is generally thought to have purely neurotoxic effects in the context of post-stroke neuroinflammation. IL-1β promotes its own release and the release of other pro-inflammatory cytokines, stimulates neuronal calcium influx thereby potentiating neuronal apoptotic cell death, contributes to the activation and proliferation of microglia and astrocytes, strongly attracts neutrophils, and induces edema and expression of cell adhesion molecules in endothelial cells. Animals deficient in IL-1β display smaller infarcts and animals given exogenous IL-1β after stroke suffer from increased neutrophil infiltration and brain dam- age. However, IL-1β also induces production of an IL-1 receptor antagonist (IL-1ra). Administration of IL-1ra in animal models of stroke results in notable reductions in brain damage. Recombinant methionylated human IL-1ra drugs, such as Anakinra (already approved for treatment of rheumatoid arthritis), are in clinical trials with early results appearing favorable. [1-4]
TNF-α
Activated microglia release TNF-α and display up-regulated TNF receptor expression suggest- ing that the actions of TNF-α are at least partially autocrine. TNF-α is also released by infiltrating macrophages and Th1 cells. Evidence is con- flicting as to the role of TNF-α after stroke as it appears to have both neurotoxic and neuropro- tective effects. On the one hand, TNF-α is known to activate microglia and astrocytes causing the release of other pro-inflammatory and cytotoxic mediators, inhibit glutamate uptake in vitro po- tentiating neuronal excitotoxicity, and stimulate endothelial cell apoptosis promoting breakdown of the BBB. On the other hand, TNF-α enhances production of neurotrophic factors, initiates repair processes in the vascular endothelium, and encourages post-stroke neural plasticity. Further complicating the picture, membrane- bound TNF-α functions differently than soluble TNF-α, and the two TNF receptors appear to have different effects when engaged by their ligand. Therefore, further investigation as to the mechanisms of TNF-α actions will be required it can progress in its development as a therapeutic target. [2,3]
Chemokines
Cytokines including IL-1β and TNF-α directly stimulate the release of chemokines in the context of cerebral ischemia. For example, activated microglia release chemokines including IL-8 (a.k.a. CXCL8), MCP-1 (a.k.a. CCL2), and MIP-1α (a.k.a. CCL3). IL-8 attracts neutrophils, which bear the cell surface IL-8 chemokine receptors, CXCR1 and CXCR2. MCP-1 and MIP-1α attract macrophages and monocytes expressing their respective chemokine receptors, CCR1 and CCR2 and 5, respectively. Chemokines not only recruit leukocytes to the lesion site, but they also participate in stimulating apoptosis and phagocytosis. Generally, chemokine levels in brain are known to increase following ischemic stroke and inhibition or deficiency of chemokines is associated with reduced brain injury. Specifically, it has been observed that animals lacking CCR2 are protected from I/R injury and blocking the neu- trophil attracting effects of IL-8 using Reparixin, a selective CXCR1 and CXCR2 inhibitor, reduces stroke damage by inhibiting leukocyte recruit- ment. [2-4]
Adhesion Molecules
Three groups of proteins function to slow down and adhere leukocytes to the endothelial surface of blood vessels and facilitate their passage across the BBB into the lesion site – Selectins, Cellular Adhesion Molecules (CAMs), and Integrins. Upon exposure to inflammatory mediators, endothelial cells first up-regulate P-Selectin, and then E-Selectin. Circulating leukocytes bind these adhesion molecules via extracellular carbohydrate residues (sialyl-Lewisx) and begin to roll over the endothelial surface. IL-1β and TNF-α also up-regulate expres- sion of ICAM-1 and VCAM-1 on endothelial cells. Interaction of these CAMs with Integrins (LFA-1, Mac-1, and CD11c each dimerized with CD18) on leukocytes facilitates stronger binding to the endothelium and stimulates extravation. Inter- ruption of this process of leukocyte infiltration has been the focus of many studies. Synthetic sialyl-Lewisx analogs, anti-ICAM-1 antibodies, ICAM-1 antisense oligonucleotides, and anti- Mac1 antibodies all result in reduced infarct size when administered shortly after stroke and two of these strategies have entered clinical trials – Enlimomab (anti-ICAM-1 antibody) and Leukarrest (anti-MAC-1 antibody). [2,4]
Matrix Metalloproteinases (MMPs)
MMPs are zinc-dependent endopeptidases that function to degrade extracellular matrix. Two MMP family members, MMP-2 and MMP-9, are known to degrade the major components of the BBB basal lamina (i.e., type IV collagen, laminin, and fibronectin) and have been implicated in ischemic stroke. Inflammatory cytokines rapidly up-regulate MMP-9 first in endothelial cells, then in neutrophils, which degranulate releasing pro and active forms of MMP-9, and finally in macrophages. MMP-9 knock-out and neutrophil degranulation suppression strategies in animal models have been shown to reduce stroke damage. Further, SB-3CT, a selective inhibitor of MMP-2 and MMP-9, appears to reduce lesion volume in animal models of stroke. Additionally, Statin drugs and Minocycline both appear to have inhibitory effects on MMPs and are known to improve outcomes in experimental animal models. [1-4]
IL-10
IL-10 is an anti-inflammatory cytokine that functions by inhibiting IL-1β and TNF-α and their respective receptors and by reducing astrocyte activation after stroke. IL-10 is produced predom- inately by activated microglia and astrocytes as well as by TLR-stimulated monocytes, Th2 cells, and Treg cells. Tregs cells are thought to exert their neuroprotective effects via IL-10 as deple- tion of Treg cells results in TNF-α elevation and intracerebroventricular delivery of IL-10 rescues animals depleted of Tregs that would otherwise show increased damage following stroke. Clini- cally, IL-10 levels become elevated in CSF and plasma after stroke, and low plasma concentra- tions in the hours after stroke are an indicator of poor outcomes. Post-stroke administration of IL- 10 either systemically or centrally reduces infarct volume in in vivo animal models. [2-4]
Conclusion
The complex interplay between resident and invading cells and their bioactive effector signaling molecules as they initially damage and later protect and repair brain tissue after acute ischemic stroke offers many opportunities for pharmacological intervention in clinically feasible therapeutic windows. Figure 1 illustrates many of these cellular and molecular interactions and strategies under investigation for improvement of stroke patient outcomes. [1-4] This review is meant to be an overview of selected aspects of neruroinflammation after acute ischemic stroke and should be taken as neither comprehensive nor definitive.

Figure 1: Illustration of selected aspects of cellular and molecular interactions involved in neuroinflammation after acute ischemic stroke. [1-4]
MD Biosciences provides animal models of stroke and biomarker analysis to assess neuroinflammation following post-ischemic stroke.
References:
-
Candelario-Jalil, E. (2009). Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Current Opinion in Investigational Drugs 10(7):644-654.
-
Ceulemans, A.-G., Zgavc, T., Kooijman, R., Hachimi-Idrissi, S., Sarre, S., and Michotte, Y. (2010). The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. Journal of Neuroinflammation 7:74.
-
Downes, C.E. and Crank, P.J. (2010). Neural injury following stroke: are Toll-like receptors the link between the immune system and the CNS? British Journal of Pharmacology 160:1872- 1888.
-
Lakhan, S.E., Kirchgessner, A., and Hofer, M. (2009). Inflammatory mechanisms in ischemic stroke: therapeutic approaches. Journal of Translational Medicine 7:97.
-
Bacigaluppi, M., Comi, G. and Hermann, D.M. (2010). Animal models of ischemic stroke. Part two: Modeling cerebral ischemia. The Open Neurology Journal 4:34-38.